

ORIGINAL PAPER
Comparative analysis of the rectal
and caecal microbial community composition and function
in adult Erhualian and Sushan pigs
1
Jiangsu Academy of Agricultural Sciences, Institute of Animal Science, Zhongling Street 50, Xuanwu, Nanjing, Jiangsu, China
Publication date: 2021-07-28
Corresponding author
B. Li
Jiangsu Academy of Agricultural Sciences, Institute of Animal Science, Zhongling Street 50, Xuanwu, Nanjing, Jiangsu, China
Jiangsu Academy of Agricultural Sciences, Institute of Animal Science, Zhongling Street 50, Xuanwu, Nanjing, Jiangsu, China
J. Anim. Feed Sci. 2021;30(3):248-259
KEYWORDS
TOPICS
ABSTRACT
Intestinal microbiota plays an important role in nutrition, metabolism
and immunity in all mammals. It is comprised of diverse populations of
bacteria and other microorganisms whose abundances are impacted by both
environmental and host genetic factors. However, the understandings of the intestinal
microbiota in different pig breeds remain largely undefined. To examine
the differences in intestinal microflora between two pig breeds with different genetic
backgrounds under the same environment, 16S rRNA gene amplification
and sequencing were performed to investigate the structural composition and
potential functions of microbial communities in rectum and caecum of Erhualian
and Sushan pigs. The results revealed that the diversity of intestinal microflora
in two pig breeds was similar, but the abundance of specific intestinal microflora
was different. At the phylum level, the dominant bacteria in caecum and rectum
of Erhualian and Sushan pigs were Firmicutes, Acidobacteria and Bacteroides,
but their expression abundance was different. Firmicutes and Bacteroidetes in
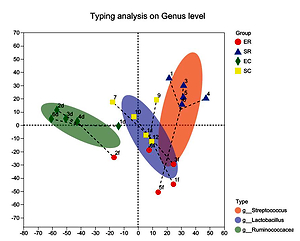
Erhualian pigs were higher than those in Sushan pigs. At the genus level, Lactobacillus
was the most abundant in caecum of Sushan pigs (6.83%) and rectum
of Erhualian pigs (9.61%), while Ruminococcaceae UCG-005 were dominant in
caecum of Erhualian pigs (10.89%) and Streptococcus in rectum of Sushan pigs
(24.89%). This study further confirmed the existence of specific microbial community
diversity and abundance in different pig breeds. The microbial community
diversity and abundance in Erhualian and Sushan pigs were closely related
to pig fat deposition and nutrient absorption.
FUNDING
This study was supported by China Agriculture
Research System of MOF and MARA.
CONFLICT OF INTEREST
The authors declare that there is no conflict of
interest.
REFERENCES (28)
1.
Bergen W.G., 2015. Small-intestinal or colonic microbiota as a potential amino acid source in animals. Amino Acids 47, 251–258, https://doi.org/10.1007/s00726....
2.
Bian G., Ma S., Zhu Z. et al., 2016. Age, introduction of solid feed and weaning are more important determinants of gut bacterial succession in piglets than breed and nursing mother as revealed by a reciprocal cross-fostering model. Environ. Microbiol. 18, 1566–1577, https://doi.org/10.1111/1462-2....
3.
Chen S., Zhou Y., Chen Y., Gu J., 2018. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890, https://doi.org/10.1093/bioinf....
4.
Choy Y.Y., Quifer-Rada P., Holstege D.M., Frese S.A., Calvert C.C., Mills D.A., La-muela-Raventos R.M., Waterhouse A.L., 2014. Phenolic metabolites and substantial microbiome changes in pig feces by ingesting grape seed proanthocyanidins. Food Funct. 5, 2298–2308, https://doi.org/10.1039/c4fo00....
5.
Diao H., Yan H.L., Xiao Y. et al., 2016. Intestinal microbiota could transfer host Gut characteristics from pigs to mice. BMC Microbiol. 16, 238, https://doi.org/10.1186/s12866....
6.
Dowarah R., Verma A.K., Agarwal N., Patel B.H.M., Singh P., 2017. Effect of swine based probiotic on performance, diarrhoea scores, intestinal microbiota and gut health of grower-finisher crossbred pigs. Livest. Sci. 195, 74–79, https://doi.org/10.1016/j.livs....
7.
Du J., Yuan Z., Ma Z., Song J., Xie X., Chen Y., 2014. KEGG-PATH: Kyoto encyclopedia of genes and genomes-based pathway analysis using a path analysis model. Mol. BioSyst. 10, 2441–2447, https://doi.org/10.1039/c4mb00....
8.
Edgar R.C., 2013. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998, https://doi.org/10.1038/nmeth.....
9.
Guo X., Xia X., Tang R., Zhou J., Zhao H., Wang K., 2008. Development of a real-time PCR method for Firmicutes and Bacteroidetes in faeces and its application to quantify intestinal population of obese and lean pigs. Lett. Appl. Microbiol. 47, 367–373, https://doi.org/10.1111/j.1472....
10.
Kemp P.F., Aller J.Y., 2004. Bacterial diversity in aquatic and other environments: what 16S rDNA libraries can tell us. FEMS Microbiol. Ecol. 47, 161–177, https://doi.org/10.1016/S0168-....
11.
Kim H.B., Borewicz K., White B.A., Singer R.S., Sreevatsan S., Tu Z.J., Isaacson R.E., 2011. Longitudinal investigation of the age-related bacterial diversity in the feces of commercial pigs. Vet. Microbiol. 153, 124–133, https://doi.org/10.1016/j.vetm....
12.
Kim H.B., Isaacson R.E., 2015. The pig gut microbial diversity: understanding the pig gut microbial ecology through the next generation high throughput sequencing. Vet. Microbiol. 177, 242–251, https://doi.org/10.1016/j.vetm....
13.
Langille M.G., Zaneveld J., Caporaso J.G. et al., 2013. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821, https://doi.org/10.1038/nbt.26....
14.
Magoč T., Salzberg S.L., 2011. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963, https://doi.org/10.1093/bioinf....
15.
Mathur R., Barlow G.M., 2015. Obesity and the microbiome. Expert Rev. Gastroenterol. Hepatol. 9, 1087–1099, https://doi.org/10.1586/174741....
16.
Niu Q., Li P., Hao S., Kim S.W., Du T., Hua J., Huang R., 2019. Characteristics of gut microbiota in sows and their relationship with apparent nutrient digestibility. Int. J. Mol. Sci. 20, 870, https://doi.org/10.3390/ijms20....
17.
Pajarillo E.A.B., Chae J.P., Balolong M.P., Kim H.B., Seo K.-S., Kang D.-K., 2014. Pyrosequencing-based analysis of fecal microbial communities in three purebred pig lines. J. Microbiol. 52, 646–651, https://doi.org/10.1007/s12275....
18.
R Core Team, 2020. R: a Language and Environment for Statistical Computing. R Foundation for Statistical Computing. Vienna (Austria), https://www.r-project.org.
19.
Robinson M.D., McCarthy D.J., Smyth G.K., 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140, https://doi.org/10.1093/bioinf....
20.
Segata N., Izard J., Waldron L., Gevers D., Miropolsky L., Garrett W.S., Huttenhower C., 2011. Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60, https://doi.org/10.1186/gb-201....
21.
Schloss P.D., Westcott S.L., Ryabin T. et al., 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541, https://doi.org/10.1128/AEM.01....
22.
Simpson J.M., McCracken V.J., White B.A., Gaskins H.R., Mackie R.I., 1999. Application of denaturant gradient gel electrophoresis for the analysis of the porcine gastrointestinal microbiota. J. Microbiol. Methods 36, 167–179, https://doi.org/10.1016/S0167-....
23.
Thaiss C.A., Zmora N., Levy M., Elinav E., 2016. The microbiome and innate immunity. Nature 535, 65–74, https://doi.org/10.1038/nature....
24.
White J.R., Nagarajan N., Pop M., 2009. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comp. Biol. 5, e1000352, https://doi.org/10.1371/journa....
25.
Woting A., Pfeiffer N., Loh G., Klaus S., Blaut M., 2014. Clostridium ramosum promotes high-fat diet-induced obesity in gnotobiotic mouse models. mBio 5, e01530-14, https://doi.org/10.1128/mBio.0....
26.
Xiao Y., Kong F., Xiang Y., Zhou W., Wang J., Yang H., Zhang G., Zhao J., 2018. Comparative biogeography of the gut microbiome between Jinhua and Landrace pigs. Sci. Rep. 8, 5985, https://doi.org/10.1038/s41598....
27.
Yang H., Xiao Y., Wang J., Xiang Y., Gong Y., Wen X., Li D., 2018. Core gut microbiota in Jinhua pigs and its correlation with strain, farm and weaning age. J. Microbiol. 56, 346–355, https://doi.org/10.1007/s12275....
28.
Yang L., Bian G., Su Y., Zhu W., 2014. Comparison of faecal microbial community of Lantang, Bama, Erhualian, Meishan, Xiaomeishan, Duroc, Landrace, and Yorkshire sows. Asian-Australas. J. Anim. Sci. 27, 898–906, https://doi.org/10.5713/ajas.2....
Share
RELATED ARTICLE
| ISSN: | 1230-1388 |
Assigning DOI numbers, introducing articles from supplements and recent publications to POL-index database, maintaining anti-plagiarism detection, electronic system to proceed manuscripts and webpage of the Journal with interactive pdf files, and language correction of manuscripts published in Journal of Animal and Feed Sciences are financed in the years 2018–2019 by the Ministry of Science and Higher Education from the funds for science popularization activities, Agreement No. 631/P-DUN/2018.

© 2006-2024 Journal hosting platform by Bentus
We process personal data collected when visiting the website. The function of obtaining information about users and their behavior is carried out by voluntarily entered information in forms and saving cookies in end devices. Data, including cookies, are used to provide services, improve the user experience and to analyze the traffic in accordance with the Privacy policy. Data are also collected and processed by Google Analytics tool (more).
You can change cookies settings in your browser. Restricted use of cookies in the browser configuration may affect some functionalities of the website.
You can change cookies settings in your browser. Restricted use of cookies in the browser configuration may affect some functionalities of the website.